

Introducing ferro-hgvs
Faster, more complete HGVS variant parsing for the whole genomics community

Every genetic variant reported in a clinical laboratory has a precise, standardized name. It looks something like NM_000249.4:c.350C>T, and what it communicates is specific: in the MLH1 gene, at position 350 of the coding sequence, a cytosine changes to a thymine. That compact string is how clinical labs communicate findings to each other, how oncology reports describe mutations, and how hundreds of millions of records in global databases like ClinVar are stored, retrieved, and compared.
This naming system is called HGVS nomenclature, named for the Human Genome Variation Society that maintains it. It is the backbone of clinical genomics, used in hereditary cancer panels, rare disease diagnosis, pharmacogenomics testing, and variant database submissions worldwide. The software tools that process HGVS strings are infrastructure that the field depends on every day, often without thinking about it.
Fulcrum Genomics is releasing ferro-hgvs (v0.1.0), a new open-source HGVS parser and normalizer written in Rust. ferro-hgvs is the fastest HGVS tool available by a wide margin, supports the broadest set of HGVS patterns of any open-source library, has been validated against millions of real clinical variants, and is accompanied by infrastructure that makes the tools you already use work better. A live web service at hgvs.acgt.bio lets you try everything described in this post with no installation required.
The HGVS Standard
The authoritative HGVS specification is maintained at hgvs-nomenclature.org by the HGVS Variant Nomenclature Committee (HVNC), a working group of the Human Genome Organization. The standard covers six coordinate systems: genomic (g.), coding DNA (c.), non-coding (n.), RNA (r.), protein (p.), and mitochondrial (m.). It defines rules for every variant type from simple substitutions to complex repeat expansions, and applies across multiple reference sequence types including NM_, NR_, NC_, NG_, LRG_, and NP_ accessions.
The standard has been evolving for decades. Beginning with version 21.0.0 in January 2024, the HVNC adopted semantic versioning and introduced formal computational grammar using Extended Backus-Naur Form (EBNF), improving both the precision of the specification and its implementability in software. A December 2024 paper in Genome Medicine (Hart et al.) summarizes these improvements.
The standard is also a living document. The HVNC manages changes through a public Community Consultation process, where proposed extensions and clarifications are published for open community comment before ratification. These proposals matter because many of them describe patterns that already appear in real clinical data, in ClinVar submissions, lab reports, and variant databases, before they are formally adopted. A tool that does not track these proposals will reject or mishandle variants that clinical teams encounter every day.
ferro-hgvs supports all ratified consultation proposals, including SVD-WG001 (reporting variants confirmed to be unchanged, such as c.1823A=, important for negative findings in clinical reporting), SVD-WG002 (non-coding DNA reference sequences with the n. prefix), SVD-WG004 (the ISCN/HGVS named extension for structural variants and chromosomal rearrangements), SVD-WG009 (the discontinuation of the conversion variant type), and the accepted proposal for circular reference sequences such as the mitochondrial genome. Beyond ratified proposals, ferro parses patterns corresponding to open consultation topics including distance-between-variants notation (SVD-WG010), handling the kinds of strings that appear in real data even where the specification is still evolving.
Why HGVS Parsing Is Harder Than It Looks
Not all variants are simple single-letter substitutions. The notation covers deletions, insertions, duplications, deletion-insertions (delins), inversions, frameshifts, tandem repeat expansions, uncertain positions, and compound variants, each with distinct rules for formatting and normalization. Protein notation adds three-letter amino acid codes, stop codon representations, extension variants, and uncertain amino acid ranges on top of that.
One of the most clinically significant challenges is intronic variant handling. Many of the most important variants in clinical genetics fall at splice sites, the boundaries between coding exons and non-coding introns, because disrupting splicing often destroys gene function entirely. These variants use an offset notation such as NM_000249.4:c.117-2del, which describes a deletion two bases before the start of exon 12 in MLH1 (a classic splice acceptor variant). Processing these correctly requires knowing not just the transcript sequence but how the transcript maps back to the genome. This is a step that most HGVS tools either skip entirely or handle only through opaque workarounds.
Normalization is a separate challenge from parsing. The same indel near a repetitive region can be written in dozens of equivalent but non-identical ways, and normalization is the process of resolving all of them to a single canonical form. This matters enormously for matching variants across databases, deduplicating records, and submitting to ClinVar, where a non-normalized form may fail submission or be treated as a novel variant. A simple example: NM_000249.4:c.1852_1853delAA normalizes to NM_000249.4:c.1852_1853del, because the explicit deleted sequence is redundant under HGVS guidelines.
Finally, HGVS strings in the real world frequently deviate from the formal specification. Clinical labs, VEP annotators, legacy databases, and manual curators have produced decades of HGVS strings that use lowercase amino acids, omit version numbers, use informal shorthand, or reflect older versions of the standard. A production tool for clinical genomics needs to handle these gracefully, with configurable behavior, rather than simply rejecting them.
The Current Tool Landscape
Three main tools are currently in wide use for HGVS parsing and normalization: mutalyzer (from Leiden University Medical Center), biocommons/hgvs (the Python reference implementation), and hgvs-rs (a Rust port of biocommons). All three are valuable contributions to the field, and ferro-hgvs builds on the work they represent.
Speed is the most visible limitation. All three tools run at roughly 20 variants per second or fewer when operating locally. In network-dependent mode, which biocommons and mutalyzer require for some operations by default, throughput drops to 0.2 to 1 variant per second. At those rates, processing a complete ClinVar export of over one million variants takes days. For real-time annotation in clinical pipelines, or for researchers running large cohort studies, this is a genuine bottleneck.
Coverage gaps compound the speed problem. The table below shows validation (V) and normalization (N) support across the four tools, as tested live at hgvs.acgt.bio. The most significant gap is intronic c. coordinate normalization, a category that covers a large fraction of clinically actionable splice-region variants. hgvs-rs can validate these but cannot normalize them. biocommons can normalize them but requires a running UTA database. mutalyzer handles them by rewriting to genomic coordinates behind the scenes. Only ferro normalizes them natively and transparently.
Table 1: HGVS Feature Support by Tool (V = validate, N = normalize, V/N = both)

Operational complexity is an additional barrier. biocommons requires a running UTA PostgreSQL database and SeqRepo, a locally stored sequence repository. Configuring and containerizing these dependencies for reproducible clinical pipelines takes significant effort. The end result is that teams often fall back to network-dependent operation, which sacrifices both speed and reproducibility.
Introducing ferro-hgvs
ferro-hgvs is written in Rust and uses nom, a well-established Rust parsing library, to implement zero-copy parsing of HGVS strings. Variants are parsed without allocating intermediate string representations, which contributes to the tool’s throughput and makes it suitable for memory-constrained environments. The parser produces a typed abstract syntax tree that covers every valid HGVS form, giving downstream code precise, structured access to every component of a variant without manual string manipulation.
The name follows Fulcrum’s growing Rust-based genomics toolchain, where “ferro” (iron in Latin and Spanish) reflects the language’s reputation for performance and reliability. This is the same philosophy that produced fgumi, Fulcrum’s Rust port of fgbio’s UMI-handling tools.
Performance
ferro-hgvs parses approximately 4 million HGVS patterns per second and normalizes approximately 2.5 million variants per second, all offline after a one-time reference data preparation step. The table below compares performance against existing tools. The speedups are large enough that they change what is computationally feasible: a normalization run over all of ClinVar that would take days with existing tools takes minutes with ferro-hgvs.
Table 2: Normalization Performance Comparison

Error Handling
ferro-hgvs provides three configurable error modes to match different workflow needs. Strict mode rejects any non-conformant input and is appropriate for validation pipelines where every variant must be checked carefully. Lenient mode auto-corrects common deviations (for example, converting the lowercase p.val600glu to the correctly capitalized p.Val600Glu) while emitting warnings, which is useful for batch processing real-world data. Silent mode applies the same auto-corrections without logging warnings, useful when processing large legacy datasets where noise from formatting issues would obscure other information.
Error and warning behavior can be tuned per-code through a .ferro.toml configuration file, and the ferro explain command provides plain-English explanations for every error and warning code, making it practical for teams to understand and act on quality issues in their variant data.
Tested Against the Real World
Performance numbers only matter if the tool is correct. ferro-hgvs has been validated against multiple large corpora of real HGVS data, and the testing strategy was designed to leave as few gaps as possible.
ClinVar and major variant databases. ferro-hgvs has been run against the full ClinVar database, over one million HGVS strings spanning decades of submissions from clinical laboratories worldwide. ClinVar contains variants from every major gene, disease area, and variant type, including many edge cases and historically inconsistent notations that accumulate in any large curated database. In addition to ClinVar, the tool has been validated against variant sets from gnomAD, LOVD (the Leiden Open Variation Database), and other major genomic resources, representing the full breadth of HGVS usage across research and clinical communities.
VEP-annotated VCFs. The Variant Effect Predictor (VEP) is the most widely used annotation tool in genomics, and its HGVS output appears in tumor sequencing reports, population studies, and research publications at scale. VEP-annotated HGVS strings frequently deviate subtly from the formal specification in ways that are predictable but that naive parsers reject. ferro-hgvs has been validated extensively against VEP outputs specifically because they represent the kind of real-world data that clinical pipelines encounter most often.
Client pipeline data. Fulcrum Genomics works with biotech and clinical genomics teams across oncology, rare disease, and pharmacogenomics. That work has produced a large internal corpus of HGVS strings from diverse laboratory platforms, annotation tools, and reporting systems, which directly informed the edge cases ferro was designed to handle. Real production data surfaces failure modes that no synthetic test suite can fully anticipate.
Borrowed test suites from competing tools. One of the most rigorous validation steps was incorporating the test suites from mutalyzer, biocommons/hgvs, and hgvs-rs directly into ferro’s testing framework. These are the carefully curated sets of specific variants, tricky edge cases, and known failure modes that each tool’s development team built and refined over years. By running ferro against all of them and checking that it produces equivalent or better results, we can confirm that ferro’s correctness is at least as strong as each competing tool in their own strongest areas. Where ferro and another tool produce different outputs, the comparison is surfaced explicitly, which is itself useful information for the community about where implementations diverge and why.
Fuzz testing. ferro-hgvs includes a fuzz testing harness that generates millions of pseudo-random HGVS-like strings to find crashes, panics, and unexpected behavior at the parser boundary. This kind of robustness testing is rare in bioinformatics software and helps ensure that ferro does not fail silently or unpredictably when it encounters malformed input, which is a common occurrence when processing data from heterogeneous clinical sources.
Better Infrastructure for the Whole Ecosystem
One of the most time-consuming aspects of running any HGVS tool locally is assembling the reference data it needs: transcript sequences, genome assemblies, transcript-to-genome mappings, and protein sequences. Getting this data, keeping it versioned, and making it available to tools in the right formats is work that every team running HGVS software has to do independently.
The ferro prepare command solves this once. It downloads and organizes the complete reference dataset needed for comprehensive HGVS normalization from authoritative sources (NCBI, EBI, and MANE), validates the downloads, and structures them in a format that ferro can use offline without any network access during normal operation. The dataset totals approximately 6 GB:
Table 3: Reference Data Prepared by ferro prepare
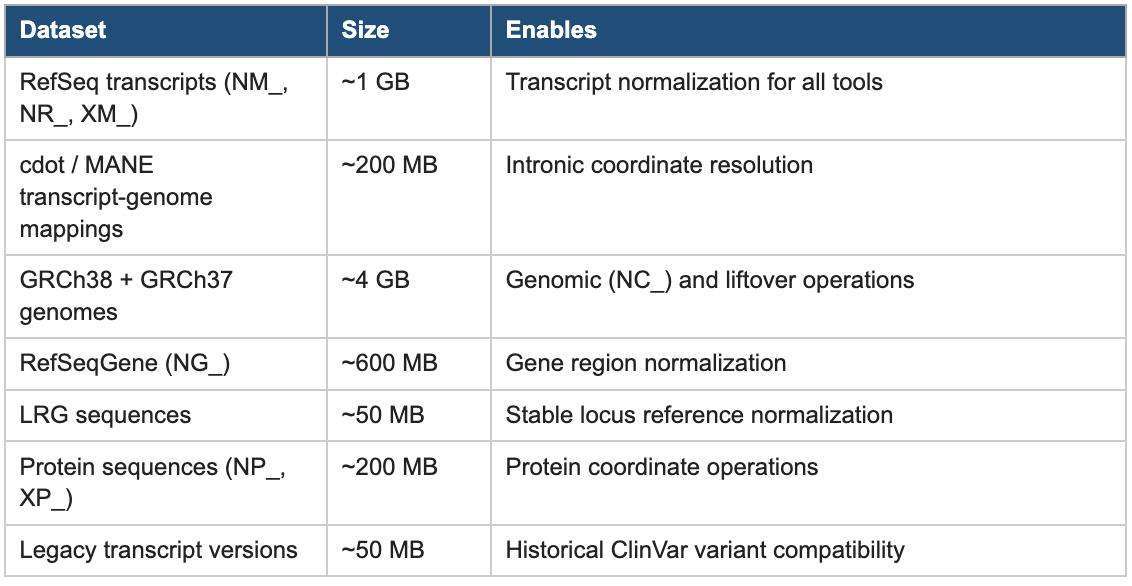
Crucially, this reference dataset is not ferro-proprietary. The same data is shared with mutalyzer, biocommons/hgvs, and hgvs-rs, enabling all of them to run fully offline with consistently versioned, high-quality reference data. Teams that continue to use existing tools can still run ferro prepare once and immediately benefit from better-quality local reference data for all of their HGVS workflows.
A related community contribution is the ferro-benchmark harness. mutalyzer, biocommons, and hgvs-rs are all single-threaded by design, which limits how quickly they can process large variant sets even on multi-core hardware. The benchmark harness parallelizes all of them across CPU cores, dramatically improving their throughput for large validation and benchmarking runs. The ferro-benchmark compare results command then surfaces disagreements between tools variant-by-variant, making tool differences visible and actionable. This infrastructure is available to anyone running tool evaluation or validation studies, regardless of whether they adopt ferro as their primary tool.
Try It Now: hgvs.acgt.bio
The fastest way to experience ferro-hgvs is the live web service at hgvs.acgt.bio. No account, no installation, and no configuration is required. Paste any HGVS variant and see results in seconds. The service is backed by ferro-hgvs and exposes its full feature set through a tabbed interface, along with a REST API for programmatic access.
Multi-tool normalization and comparison. The normalize tab runs any HGVS string through ferro, mutalyzer, biocommons, and hgvs-rs simultaneously and displays their outputs side by side. The service detects agreement and disagreement automatically, shows processing time per tool, and provides a detailed component breakdown from ferro showing reference, coordinate system, variant type, position, and whether shifting occurred during normalization. Batch mode accepts a list of variants for processing all at once. This is a useful diagnostic for teams evaluating tools or investigating why a variant is producing inconsistent results across systems.
Coordinate conversion. The convert tab translates variants between coordinate systems: c. to g., g. to c., c. to p., and n. to g. Provide a coding variant like NM_000249.4:c.350C>T and retrieve the equivalent genomic position on GRCh38, or calculate the protein consequence. An option to return all conversions across all mapped transcripts at once is available for variants where multiple transcripts are relevant.
Variant effect prediction. The effect tab predicts variant consequences using standardized Sequence Ontology (SO) terms: splice_site_variant, frameshift_variant, inframe_deletion, missense_variant, stop_gained, and others. Each prediction includes an impact level (HIGH, MODERATE, LOW, or MODIFIER) and optionally a prediction of whether a truncating variant would trigger nonsense-mediated mRNA decay (NMD), a key factor in determining whether a premature stop codon produces a loss-of-function outcome.
Genomic liftover. The liftover tab converts coordinates between GRCh37 (hg19) and GRCh38 (hg38) in either direction. It accepts both raw chromosome positions (chr7:117120148) and HGVS genomic notation (NC_000007.13:g.117120148) and returns the lifted position, the equivalent HGVS g. string, and the relevant chain region.
VCF conversion. The VCF tab handles bidirectional conversion between HGVS and VCF format. Convert a genomic HGVS variant to VCF fields (CHROM, POS, REF, ALT), or provide VCF fields and optionally a transcript accession to receive the g., c., and p. HGVS representations in a single step.
REST API. Every operation is also available via a REST API for integration into existing pipelines: POST /api/v1/normalize, /api/v1/batch/normalize, /api/v1/convert, /api/v1/effect, /api/v1/liftover, /api/v1/hgvs-to-vcf, and /api/v1/vcf-to-hgvs. All endpoints return JSON with results, processing time, and any errors or warnings. A /api/v1/validate endpoint runs ferro-only syntax checking without requiring reference data. The /health/detailed endpoint exposes the live status of all four tools and a pass/fail matrix for a built-in set of test variants.
A good starting point is to paste NM_000249.4:c.117-2del (an MLH1 splice acceptor deletion) into the normalize tab with all four tools selected. The results make the coverage difference between tools immediately visible.
Getting Started
Bioconda (recommended for most users). A Bioconda recipe is in review (PR #62795) and will be available shortly. Once merged, installation is:
conda install -c bioconda ferro-hgvs
The Bioconda build includes the benchmark and hgvs-rs comparison features. After installation, run ferro prepare --output-dir ferro-reference once to download reference data, then ferro normalize to start processing variants.
Rust library. Add to your Cargo.toml:
ferro-hgvs = “0.1”
CLI (via cargo). cargo install ferro-hgvs installs the ferro binary directly.
Python. A Python package backed by PyO3 bindings is available via pip install ferro-hgvs, providing Rust-level performance from Python code.
Web service. hgvs.acgt.bio requires no installation at all and is the quickest way to evaluate the tool.
Current Status and What Is Coming
ferro-hgvs v0.1.0 is alpha software. The parser and normalizer have been extensively validated against real clinical data, but the API is not yet stable and should be expected to evolve. The alpha designation reflects the early stage of the project, not the level of testing behind it.
Known gaps in the current release: protein (p.) and RNA (r.) normalization are validate-only in v0.1.0, repeat expansion normalization is not yet implemented, and Ensembl (ENST) transcript accessions are not supported. All of these are on the roadmap. p. and r. normalization and ENST support are the near-term priorities.
Issue reports and pull requests are welcome at the ferro-hgvs GitHub repository. Real-world HGVS strings that produce unexpected results are among the most valuable contributions the community can make. Contact Fulcrum Genomics to discuss production deployments, custom integrations, or sponsoring specific feature development.
Closing Thoughts
A variant name is only as useful as the software that can reliably parse, normalize, and compare it. When tools disagree with each other, or when they quietly fail on patterns that appear constantly in clinical data, that uncertainty propagates through pipelines, reports, and databases in ways that are hard to detect and harder to correct.
ferro-hgvs is built against the living HGVS specification at hgvs-nomenclature.org, validated on the largest and most diverse HGVS corpora we could assemble, and designed to raise the floor for the whole ecosystem rather than just replace individual tools. We hope it is useful to the community and look forward to hearing how people are using it.
Start with hgvs.acgt.bio. Paste a variant you care about. See what all four tools say.
Resources
Web service: hgvs.acgt.bio
GitHub: github.com/fulcrumgenomics/ferro-hgvs
Rust crate: crates.io/crates/ferro-hgvs
API docs: docs.rs/ferro-hgvs
HGVS specification: hgvs-nomenclature.org/stable
Community consultation proposals: hgvs-nomenclature.org/stable/consultation
Fulcrum Genomics: fulcrumgenomics.com
Fulcrum Genomics is a bioinformatics consulting firm built by scientists at the forefront of large-scale genomic research, with deep expertise in sequencing technology, pipeline engineering, and genomic data analysis for biotech, pharma, and academia. Engage us through project-based work, fractional R&D, or hourly consulting. Contact us to discuss your project.