

Introducing fgumi
A New UMI Toolkit for Next-Gen Sequencing

Years ago, when UMIs were becoming standard in high-accuracy sequencing workflows, we (Fulcrum co-founders Tim Fennell and Nils Homer) built fgbio to handle extraction, grouping, consensus calling, and the mechanics required to make error-corrected sequencing practical. It became widely adopted across research and clinical pipelines, and we’ve maintained it ever since.
And eventually, we ran into its limits.
Amplicon panels grew. Some assays began stacking hundreds of UMIs at a single locus with thousands of reads in a group. Error-corrected sequencing moved beyond small panels into exomes and other much larger datasets.
Tim describes what that looked like in practice:
“When dealing with amplicon sequencing with hundreds of UMIs at the same location and many thousands of reads, runtime just explodes. Some of the algorithms you’d like to use become infeasible.”
The underlying issue was straightforward. fgbio is single-threaded. It was written at a time when that design choice wasn’t a major limitation.
As Nils puts it:
“Error-corrected sequencing used to run on small targeted panels, and fgbio could handle that in minutes. As the field moved to exomes, those same tools went from minutes to hours.”
Around the same time, Tim and Nils were seeing accelerated UMI tools appear in commercial products, often referencing fgbio’s behavior.
“If anyone was going to build a faster implementation,” Tim says, “we felt like it should probably be us.”
That work became fgumi.
What fgumi focuses on
fgumi covers the core pieces of a UMI workflow:
Extract UMIs directly from FASTQ files
Group reads by UMI
UMI-aware deduplication
Consensus calling (simplex, duplex, and CODEC)
Filtering and metrics generation
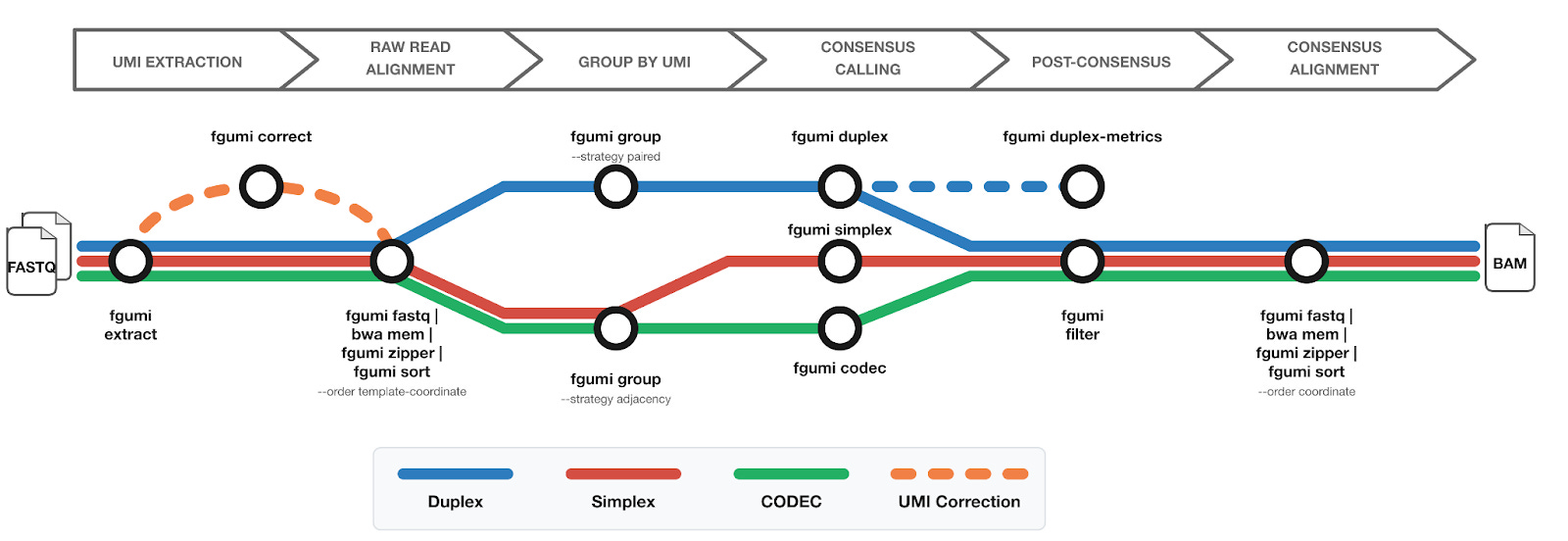
The overall structure will look familiar if you’ve worked with fgbio or pipelines like nf-core/fastquorum.
The intention was to preserve the way these pipelines already operate.
Tim explains the constraint that guided most design decisions:
“It really needed to be a drop-in replacement for fgbio for the main workflows. If moving to fgumi required a lot of work or changed how the tools behaved, adoption would be hard.”
Nils frames the priorities this way:
“Equivalency first, performance second, scope third.”
Each tool in fgumi is expected to produce the same results as the corresponding tool in fgbio. During development, this comparison surfaced small areas of non-determinism in fgbio itself, which were corrected there as well.
After outputs matched, performance work began in earnest. Rewriting in Rust allowed multi-threading and more direct control over memory usage and concurrency.
The project also kept a fairly tight scope.
“fgbio does a lot of things beyond UMI processing,” Nils says. “fgumi focuses on the core UMI tools and nothing else.”
Even sorting functionality was implemented directly so that the full processing path could be controlled and tuned.
On our deepest benchmark dataset, the full consensus pipeline, from extraction through consensus calling and filtering, completes in about 70 seconds compared to 30 minutes in fgbio, roughly a 25x speedup.
Building the rewrite
fgbio was built incrementally over many years, with Tim and Nils reviewing essentially every line of code.
fgumi was developed differently.
Some of the development work was done with the help of AI-assisted coding tools. Reviewing every line at the same level wasn’t practical at that pace.
Nils describes the adjustment:
“Letting go of reviewing every line was probably the hardest tradeoff.”
Confidence instead came from testing and validation.
fgbio itself served as the reference implementation. The large unit-test suite that had accumulated over the years was ported across. Automated review tools checked pull requests, and vendor partners provided real assay datasets for validation.
“The safety net was deep,” Nils says. “fgbio was always there as a reference, and the tests tell you whether the outputs match.”
What this should enable
The immediate impact of fgumi is runtime. Multi-threading and implementation changes make a large difference when grouping and consensus calling have to operate across very deep datasets.
That matters for several reasons.
Clinical pipelines often rely on error-corrected sequencing and have tighter turnaround requirements. Faster UMI processing also reduces compute cost when running large numbers of samples. And certain research workflows, such as large ecDNA studies, become easier to scale.
Looking further ahead, Nils hopes the practical limitations simply fade away:
“Five years from now, nobody should have to think twice about running error-corrected sequencing on a whole genome at ultra-deep coverage. The answer should just come back fast, and it should be right.”
Current status
fgumi is currently in alpha. The toolkit is functional and is being tested across a variety of vendor-provided datasets.
The team is targeting June 1, 2026 as the point when fgumi can be recommended over fgbio for production use.
We’ve also published documentation that includes:
A best practice pipeline from FASTQ to filtered consensus reads
A performance tuning guide for threading and memory
Tools for simulating UMI data and for comparing output files
The code is open source under the MIT license.
fgumi grows directly out of the same work that produced fgbio: building and maintaining UMI workflows in real sequencing projects.
If you’re already using UMI-based sequencing in your workflows, we encourage you to take a look at fgumi and see how it fits your use cases. The code is open source, the documentation is available, and your feedback will help guide the project as it matures toward production readiness.
🔗 https://github.com/fulcrumgenomics/fgumi
Fulcrum Genomics is a bioinformatics consulting firm built by scientists at the forefront of large-scale genomic research, with deep expertise in sequencing technology, pipeline engineering, and genomic data analysis for biotech, pharma, and academia. Engage us through project-based work, fractional R&D, or hourly consulting. Contact us to discuss your project.