

fastquorum: Making UMI consensus boring (in the best way)
If you’ve worked with UMI-tagged sequencing data long enough, you already know the theory. Tag molecules early, group reads that came from the same original fragment, collapse them into a consensus, and dramatically reduce error. In theory, it’s straightforward.
In practice, it’s rarely clean.
UMI consensus workflows tend to grow organically: a script here to extract tags, another to sort and group reads, a carefully tuned command copied from a past project, plus a handful of assumptions that only live in someone’s head. The science is sound, but the implementation is fragile. Small changes in library structure, read layout, or scale can quietly break things. Or worse, corrupt results without anyone noticing.
That’s the gap fastquorum was built to close.
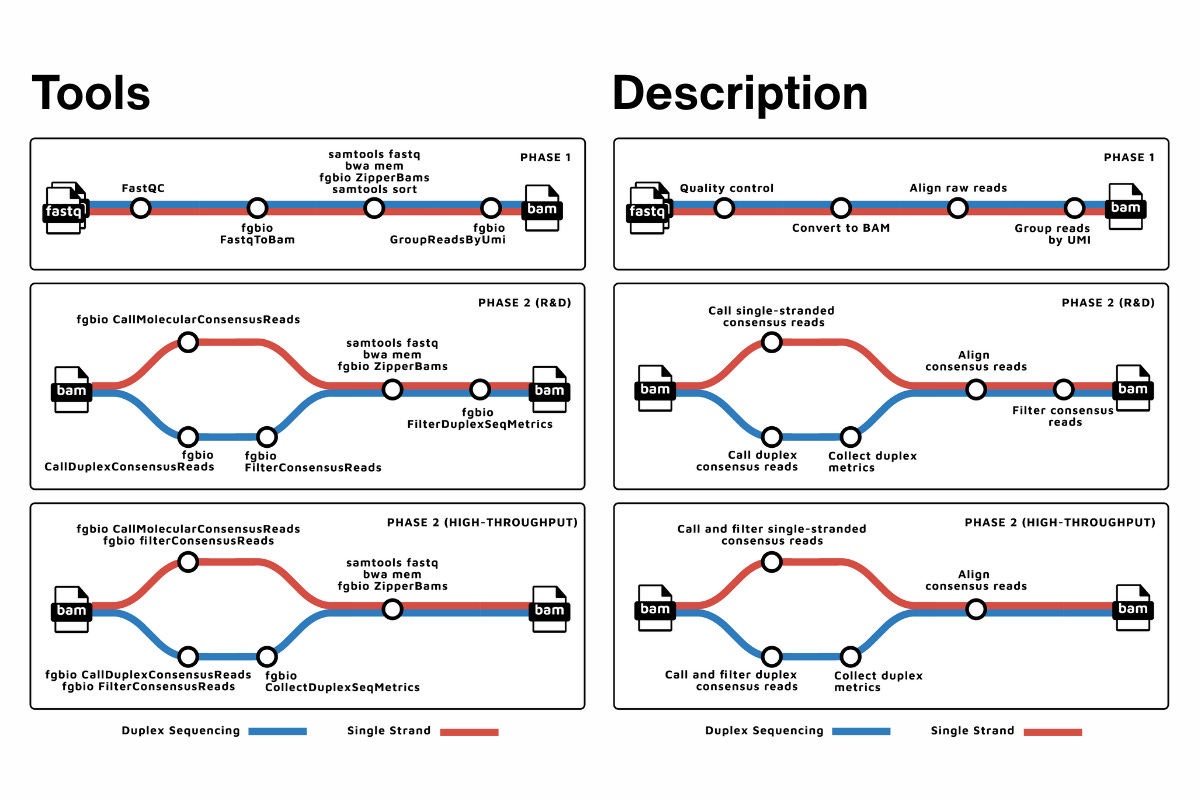
fastquorum is an nf-core pipeline built by Fulcrum Genomics that implements the fgbio FASTQ-to-consensus best-practice workflow in a way that’s reproducible, inspectable, and boringly consistent. It doesn’t invent a new approach to UMI consensus. It takes an approach many teams already intend to use and makes it reliable enough to trust across projects, people, and environments.
From “works once” to “works every time”
At a high level, fastquorum does exactly what you’d expect: it takes raw FASTQs, extracts UMIs, aligns reads, groups them by molecular family, and generates single-strand or duplex consensus reads. Quality metrics are captured along the way so you can see what’s happening, not just accept a final BAM on faith.
What matters more than the individual steps is that those steps are locked into a tested, versioned workflow. fastquorum runs under Nextflow using the nf-core framework, which means the same pipeline can be executed on a laptop, an HPC cluster, or in the cloud, with the same logic, the same tool versions, and the same outputs.
If that sounds mundane… It’s not.
Reproducibility is often treated as a compliance box to check, but in UMI-based workflows it’s foundational. Consensus calling is sensitive to grouping rules, filtering thresholds, and read handling details. When those details drift between runs or between analysts, you can end up “discovering” biology that’s really just pipeline variance.
fastquorum removes that variable.
What it’s good at — and what it isn’t
fastquorum shines when UMI consensus is a means to an end, not the end itself. If you’re doing rare variant detection, duplex sequencing, or any application where error suppression is table stakes, fastquorum gives you a clean, standardized starting point for downstream analysis.
It’s especially useful when:
Multiple projects or teams need to process UMI data the same way
You want to compare results across runs without second-guessing the pipeline
You’re tired of maintaining bespoke glue code around tools
But it doesn’t magically fix experimental limitations. Systematic sequencing errors, biased library prep, or insufficient UMI diversity will still show up in the data. fastquorum won’t hide those problems, and that’s a feature, not a bug.
Why this fits in Fulcrum’s UMI toolset
Like MutSeqR, fastquorum reflects our belief that the hardest problems aren’t caused by missing algorithms, but by brittle implementations of well-understood methods.
By packaging UMI consensus into a transparent, reproducible workflow, fastquorum makes it easier for teams to focus on interpretation, validation, and decision-making instead of debugging the same pipeline assumptions over and over again.
If UMI consensus is part of your workflow today, fastquorum is worth a look. It lets this part of your analysis fade into the background, exactly where infrastructure should be.
💻 Access the code: https://github.com/nf-core/fastquorum/tree/1.2.0
Fulcrum Genomics is a bioinformatics consulting firm built by scientists at the forefront of large-scale genomic research, with deep expertise in sequencing technology, pipeline engineering, and genomic data analysis for biotech, pharma, and academia. Engage us through project-based work, fractional R&D, or hourly consulting. Contact us to discuss your project.